
































Sulfuric acid is an important chemical with applications across different industries including crude oil refining, agricultural and pharmaceutical chemical production, and hydrometallurgical treatment. The strong corrosiveness of sulfuric acid means that selection of materials used in the equipment handling sulfuric acid requires careful consideration of its corrosion characteristics. Across the wide range of applications, sulfuric acid is mixed with diverse chemicals which can potentially alter its corrosivity. In the absence of oxidizing species, such as salts, sulfuric acid behaves as a reducing environment across the entire concentration regime. And when mixed with other reducing acids, such HCl and HF, the mixture behaves as reducing environments as well. While HCl accelerates corrosion when mixed with sulfuric acid, HF accelerates corrosion at low H₂SO₄ concentration but inhibits corrosion at high H₂SO₄ concentration. The purpose of this post is to understand the different corrosion tendencies in these solutions and examine the ability of the OLI Corrosion model to explain and predict corrosion properties when applied to these systems.
Understanding the reason for the different tendencies of HCl and HF mixtures and quantitatively predicting the corrosion characteristics in any given solution composition requires a general model that is mechanistic in nature. It needs to incorporate the complex interplay between the effect of solution thermodynamics, kinetics, transport phenomena, and alloy composition on corrosion. The key components of the OLI Corrosion model is a mixed-potential model that describes the anodic and cathodic reactions and a speciation-based thermodynamic model that describes phase and chemical equilibria (for more information on the OLI Corrosion model V11 see here). These features help the model accurately describe the speciation in solution and its effect on the electrochemical behavior of alloys.
Speciation and corrosion rates: H₂O + H₂SO₄ + HCl/HF
In concentrated sulfuric acid solution, the redox potential (measured with platinized Pt) is very high. The model shows that this is the result of low activity of water and hydronium ions. The hydronium reduction reaction controls the redox potential up to about 80 wt.% H₂SO₄. Beyond this concentration, other sulfur species may participate in determining the redox potential, but their concentrations are exceedingly low, limiting their kinetics. Because it is dominated by hydronium ion reduction, sulfuric acid redox potential is consistent with it being a nonoxidizing environment essentially across the whole concentration range. The corrosion is thus driven by active dissolution and not determined by passive film formation. The parameters that govern corrosion kinetics under open-circuit conditions are the active dissolution kinetics and cathodic reduction kinetics of hydronium ions.
HCl speciates as dissociated Cl⁻ and undissociated HCl⁰ with the ratio of Cl⁻ to HCl⁰ decreasing with increasing HCl concentration. When mixed with H₂SO₄, Cl⁻ to HCl⁰ ratio decreases with increasing H₂SO₄ concentration. The corrosion rate in HCl + H₂SO₄ solution follows a similar trend as Cl⁻ ions suggesting the importance of Cl⁻ ion adsorption at the interface. HF on the other hand, is dominated by undissociated HF⁰ in pure HF and dilute HF + H₂SO₄ solution. However, fluorosulfonic acid (HSO₃F) and fluorosulfonate ion (SO₃F-) become more important at higher H₂SO₄ concentration. The model shows that the corrosion rate decreases significantly as the fluorosulfonic species concentration increases. A possible explanation for corrosion rate being high in dilute H₂SO₄ solution and low in concentrated H₂SO₄ solution is that the adsorption of fluorosulfonic species on stainless steel surface is not as strong as the HF species. For a more rigorous description of the speciation and corrosion prediction see here¹. (Note that the thermodynamic model in the paper linked here is the Mixed-Solvent Electrolyte (MSE) model, which is currently used for speciation, but not corrosion kinetics calculations).
In the above study, an alloy equivalent number defined as (Ni⁰𐤟⁷ – Cr⁰𐤟¹ + Mo + 0.5W) was developed for stainless steels and Ni-based alloys to represent corrosion behavior in H₂SO₄ + HCl and H₂SO₄ + HF mixtures. The corrosion rate did not correlate with the traditional pitting resistance equivalent number as Fe-based alloys with the same PREN as Ni-based alloys corroded at a considerably higher rate. The linear dependence of corrosion rate in both HCl + H₂SO₄ and HF + H₂SO₄ solutions is shown in figure 1. The exponent on nickel indicates the importance of Ni in imparting corrosion resistance in these environments and conversely, the detrimental effect of iron. The negative coefficient for chromium indicates that in nonoxidizing environments, Cr may be slightly detrimental. The addition of both molybdenum and tungsten are beneficial to varying degrees.
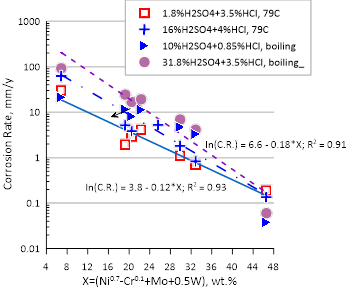

Figure 1. Effect of alloying elements on the corrosion rate of Ni-base alloys in selected (a) H₂SO₄ +HCl and (b) H₂SO₄ + HF mixtures at different conditions. Corrosion rate has been calculated using the Corrosion Analyzer and shows a linear dependence on the equivalent number defined as Ni⁰𐤟⁷ – Cr⁰𐤟¹ + Mo + 0.5W. Figure taken from Sridhar and Anderko¹.
The corrosion described until now has been in the active state. The ability of the model to describe passive dissolution is tested in the last segment. Alloy 304 in H₂SO₄ and NaCl undergoes corrosion in the active state; but under anodic protection its corrosion rate reduces significantly, and it is in the passive state.
Anodic protection of alloy 304 in an aqueous solution of H₂SO₄ and NaCl
Figure 2 shows corrosion rate data varying with the concentration of sodium chloride for alloy 304 in two different concentrations of sulfuric acid. The square symbols are the experimental corrosion rates of unprotected samples, and the triangles are the rates when the alloy is under anodic protection. Two important observations here are: 1) corrosion rate increases with increasing sulfuric acid concentration and 2) anodic protection significantly reduces corrosion. The dependence on NaCl does not follow a systematic trend due to the scatter in the data. The curves are prediction from the model; the solid curves are predictions for the unprotected alloy whereas the dashed curves are the passive current density (read off the vertical portion of the polarization curve that signifies passivity). In anodic protection, the metal is protected from corrosion by making it the anode of an electrochemical cell and holding its potential in the passive region using an external potentiostat. The passive current density is thus proportional to the corrosion rate of the alloy under anodic protection and can be used as a measure of corrosion under anodic protection. As shown in the figure, the model predictions show the dependence of corrosion rate on sulfuric acid concentration as well as the effect of anodic protection (a reduction of several orders of magnitude) within experimental scatter.
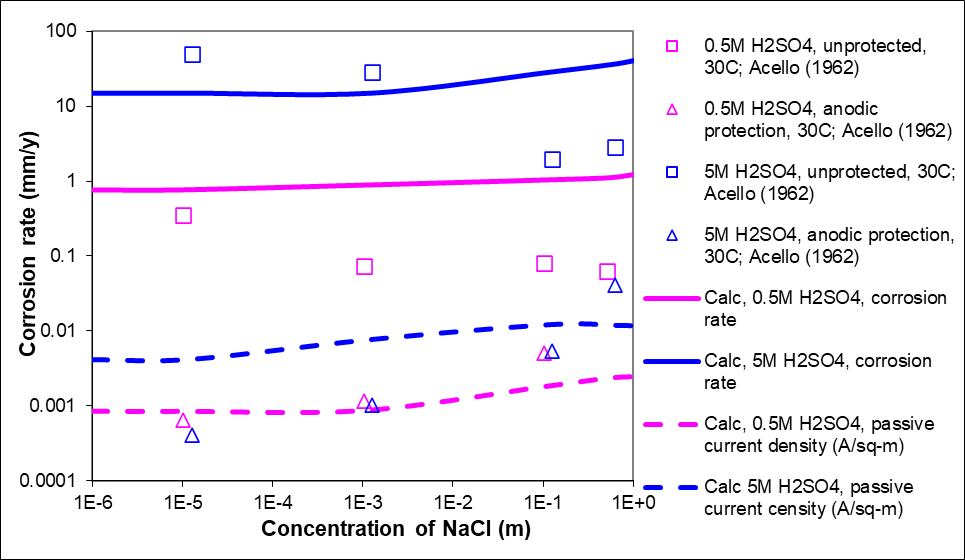
The reduction in corrosion rate by several orders of magnitude when under anodic protection is shown. The experimental data is from Acello and Greene² and the curves are model predictions from the OLI Corrosion model.
The model is thus able to predict the dissolution of alloy in both the active and passive states. The model has been extensively tested in single and mixed acid systems. And though here we focus on nonoxidizing conditions, the model reliably predicts corrosion rates in both oxidizing and nonoxidizing conditions.
References
- Sridhar, N., & Anderko, A. (2021). “Electrolyte Based Modeling of Corrosion Processes in Sulfuric Acid Mixtures, Part 1: Nonoxidizing Conditions”. Corrosion, 77(9), 935-948.
- Acello, S. J., & D. Greene, N. (1962). “Anodic protection of austenitic stainless steels in sulfuric acid-chloride media.” Corrosion, 18(8), 286t-290t.